Other Annotations
RNA-seq of K562
detail numbers
#{Gene | Total } = 59526
#{Gene | TPM==0} = 33608
#{Gene | TPM >0} = 25918
#{Gene | TPM >1} = 13184 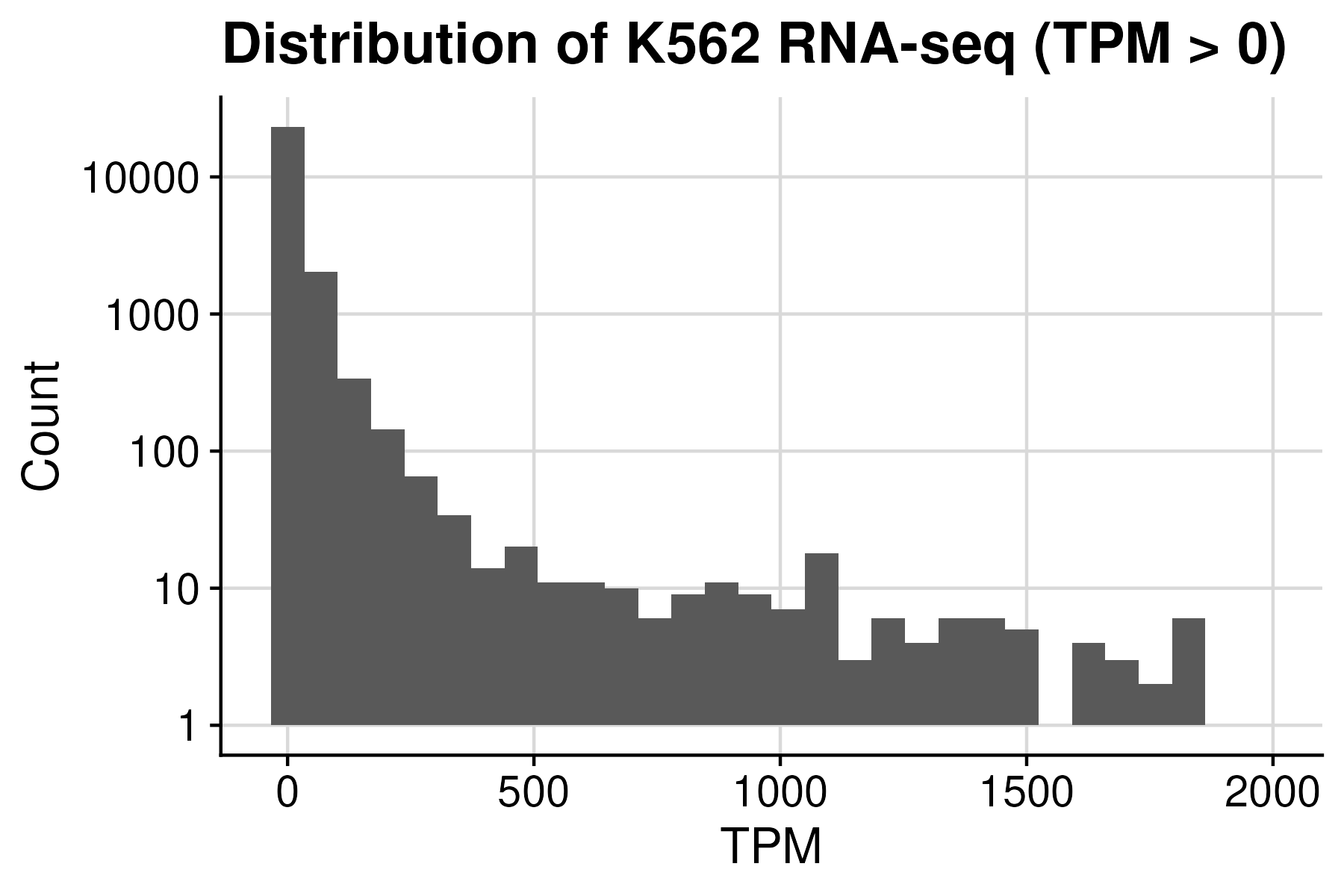
Define transcription start sites (TSS) in K562
- Select isoforms with the highest K562 Pol2 signals
- For each gene in NCBI Refgene, the isoform with highest level of pol2 chip seq (ENCFF914WIS.bigWig) at [TSS-500, TSS+500] were selected.
- If there are genes with multiple lines, it means they had isoforms with the same pol2 signal (they are mostly genes with very low pol2 signals, e,g., TP53TG3B)
- This was done by Jin-Woo. For more details, see the followin link
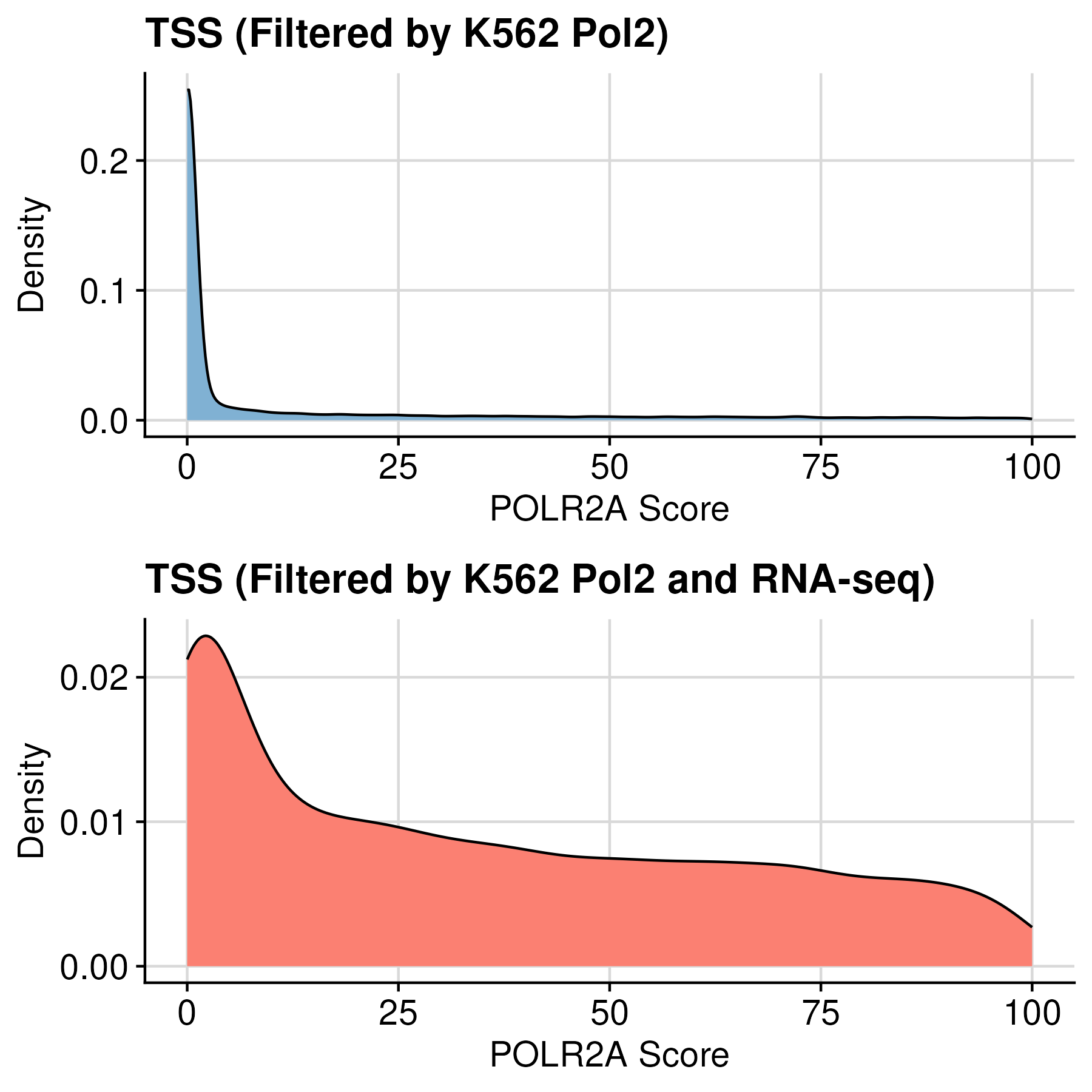
After filtering, the number of TSS = 29,330
- Filter based on RNA-seq to get K562 specific TSS
We can see that this number is still pretty high considering the number of genes expressed in a human cell type. Previous study estimated that the number of genes expressed in most human and most tissues would be around 11-13k genes, or 60-70% of refseq protein-coding genes (Ramsköld et al. 2009).
To acquire K562-specific TSS, we further filtered out lowly expressed genes using K562 RNA-seq. Applying the threshold of 1.0 TPM resulted in TSS of almost 12k genes.
ENCODE: ENCSR615EEK
Assay: RNA-seq (total RNA-seq)
Biosample: K562
Platform: Illumina NovaSeq 6000“…… Applying the threshold 0.3 RPKM, the number of genes expressed in most human and mouse tissues varied from 11,000 to 13,000, corresponding to roughly 60–70% of RefSeq protein-coding genes (Table 1).” (Ramsköld et al. 2009)
For current state, Alex proposed further selecting the TSS with corresponding gene expressed in K562 before we are able to get the cell specific TSS from the ENCODE RNA group
ENCSR615EEK – ENCODE
Assay: RNA-seq (total RNA-seq)
Biosample: K562
Platform: Illumina NovaSeq 6000Only genes with TPM larger than the threshold are left.
NUM_TPM_CUTOFF = 1
dat = dat %>% dplyr::filter(TPM > NUM_TPM_CUTOFF)detail numbers
#{TSS | NCBI RefSeq + K562 Pol2} = 29,330
=> #{TSS | NCBI RefSeq + K562 Pol2 + RNA-seq} = 11,892Download K562 TSS used in this documentation
Essential gene
DepMap: The Cancer Dependency Map project aims to identify cancer vulnerabilities by systematically profiling cancer cell lines and determining which genes are essential for their survival.
Achilles: Project Achilles, a part of DepMap, focuses on identifying and cataloging gene essentiality in cancer cell lines using techniques like shRNA and CRISPR-Cas9.
AchillesCommonEssentialControls: These are genes that are commonly essential across many cancer cell lines, used as positive controls in Project Achilles to assess the quality and reliability of the data. They help researchers ensure that the experimental setup is working as expected and that essential genes are behaving as predicted.
note:
AchillesCommonEssentialControls specifically from the second quarter of 2024 (v24Q2) data release
This list is used for quality control and normalization purposes in the Achilles pipeline, as common essential genes are expected to show consistent dependency across cell lines.
demap.v24Q2.AchillesCommonEssentialControls.csv
Total: 1,247 genes